Frequently Asked Questions¶
General¶
How should I cite GenX?¶
Give a reference to the paper describing GenX 3: A. Glavic and M. Björck J. Appl. Cryst. 55, 1063-1071 (2022) And if you want to give a link to the homepage: <http://genx.sf.net>
Issue with editing the grid on Mac¶
If you use a Swedish decimal comma then there are difficulties editing the grid. Swap to a decimal point instead in settings.
Fitting¶
Can’t fit and FOM shows inf or Nan¶
If you are using a figure of merit that uses log, for example the log fom (figure of merit) which is the standard, you probably have a negative or zero data point. Remove the data point or change the fom to a more suitable one. The same can happen if you fit with a fom that uses errors as weight and you have an error of zero somewhere.
An error message appears when I start fitting or the FOM plot and the FOM message in the toolbar differ¶
The following message appears: ERROR The disagreement between two subsequent evaluations is larger than [Number(1e-10)]. Check the model for circular assignments. In older versions this showed up as different FOM’s in the toolbar and the FOM plot.
Then you have somehow made a logical error in your model script. This means that the value of the FOM depends
on the history of the parameters. This is because when fitting the fom is evaluated for the entire population and
then at the end of each generation the program will do one additional simulation to calculate the data shown in the
gui. The plotted fom shown in the FOM folder comes from the population but the FOM that comes from the simulation
is shown in the tool bar! This could occur, for example, if you set a value of a variable that you think that you
will fit but then leave unchanged. For example (cp is a custom parameter)
def Sim(data):
....
....
cp.test = cp.test - 1.0
...
...
# Simulate something that uses cp.test
This will work all fine if you simulate your model it will behave as expected, even if you just evaluate
it. However if you fit it and leave it as a non-fitable parameter or remove it from the grid strange things will
happen!. For each evaluation the parameter cp.test will be decreased by 1.0 since it will not be given a value
from the fitting algorithm anymore. So write the Sim function like this instead:
def Sim(data):
....
....
new_test = cp.test - 1.0
...
...
# Simulate something that uses new_test
Warning
And remember never ever set a parameter so you create a circular behavior as shown above.
Note that this could also occur in a perfectly right model if the underlying computer/code has a too low
precision compared to the threshold value. You can then change the threshold value in genx.conf, section solver
parameter allowed fom discrepancy to a larger value.
Reflectivity¶
How do I add a 2Theta offset¶
Sometimes during fitting one needs to add a small 2Theta offset due to misalignment. To do this you start by
adding a custom variable in the reflectivity plugin. This is the blue nut button in the simulation tab. Lets call
the parameter TthOff.
Note
This way is deprecated since version 2.2.0 as it can now been done through the gui with the reflectivity plugin in the simulation tab. That is the Sim function does not have to be changed manually.
Then the Sim function have to be changed to something like (located in the Script tab):
def Sim(data):
I = []
# Things that should be inserted begin:
tmp0 = data[0].x*1.0
data[0].x = tmp0 + cp.TthOff
# End insertion
# BEGIN Dataset 0 DO NOT CHANGE
I.append(sample.SimSpecular(data[0].x, inst))
# END Dataset 0
# Things that should be inserted begin:
data[0].x = tmp0
# End insertion
Note that it is important to place the code outside the part marked with DO NOT CHANGE. The offset variable has been implemented in the spec_nx model.
What about different scattering length databases?¶
GenX has currently three different scattering length databases fp, fw, bc and f. The databases beginning with f relates to x-ray scattering. These are based on the Henke tables and can be found at the CXRO’s homepage. The difference between fp and fw is the scaling fp has units of electrons (or rather Thompson scattering lengths), this means that any densities in inserted in a layer using fp has to be in atoms/AA3. If one uses fw the scattering length of an atom has been scaled by its atomic weight consequently the density can be inserted in units of g/cm3. Note that the composition has to be given in weight percent of the different constituents an a compound. Both fp and fw are the scattering factors at \(Q=0\) (forward scattering) if one wants to use the Q varying atomic scattering factor the f library should be used which returns f as a function of Q. The data is collected from ESRF’s DABAX library. bc is the neutron coherent scattering length used for neutron reflectivity calculations. It has a corresponding bw which works as for fw. Note that bc is given in fm (fermi meters).
What happens if I fit the scattering length?¶
As GenX only can fit real numbers the complex part of the scattering length will be ignored.
Thus the absorption is put to zero. If you want to fit both parameters you have to make a custom variable
to scale both real and imaginary part, fit material density or choose the individual Real and Imag
variables available for fitting (e.g. Layer.setFreal/Layer.setFimag).
Could you explain how the density is defined/works with an example?¶
The key thing to understand is that the calculations use the scattering length density, the scattering length multiplied with the density. Thus, how you define and scale your scattering length must be taken into account when you define the density.
Example 1: Pure Fe. You define the scattering length as fp.Fe Then the density becomes (1 formula unit = 1 Fe atom,
Fe bcc 2 atoms/unit cell, a = 2.866): 2/2.866**3 You could also define the scattering length as one
unit cell of Fe 2*fp.Fe Then the density becomes: 1/2.866**3
Example 2: SrTiO3. You define the scattering length as fp.Sr + fp.Ti + fp.O*3 Then the density becomes
(1 formula unit = 1 unitcell of SrTiO:sub:3, a = 3.9045): 1/3.9045**3
To more easily define materials consider using the SimpleLayer plugin, which allows to calculate the SLDs from various materials parameters and automatically sets Layer x-ray and neutron parameters as well as name and density.
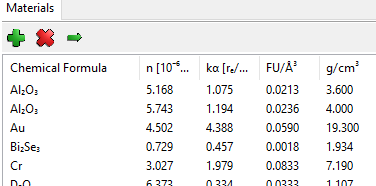
How do I transform from g/cm3 to formula unit/Å3 ?¶
I have the density of my material in g/cm3 or kg/m3, how do I use it in GenX? There are two ways: The first involves only to work with compositions in weight percent and use the fw scattering factors which are scattering lengths per weight and use densities in g/cm3. The other is recalculate the density in g/cm3 to formula units per Å3. This goes as follows:
The relative atomic mass, u, is defined as \(1 u = 1.66054\times 10-27\, \mathrm{kg}\). \(1 A = 10^-10 m\). This means that
Thus, the density (scatterers per unit volume (density in GenX)) becomes:
where
and the scattering length is written as
Let us use SrTiO3 as example, it has a density \(\rho = 5.12 g/cm^3\). The scattering length is
defined as: f = 1*fp.Sr + 1*fp.Ti + 3*fp.O. The weight of one “formula unit” becomes
\(uscatt = 1 \times 87.62 + 1 \times 47.87 + 3 \times 16.00 = 183.24\).
The density becomes: \(dens = \frac{5.12 \times 10^3}{(1.66054 \times 10^3 \times 183.24} = 0.017\)
Error when simulating spin flip reflectivity¶
I get an error when the program tries to calculate neutron spin flip reflectivity. The error is following:
It was not possible to evaluate the model script.
Check the Sim function.
... LOTS OF TEXT ...
LinAlgError: Singular matrix
This is because there is a singular matrix calculation for an incident angle of 0 or Q = 0. Try to remove the first data points and it should work.
Is it possible to automatically normalise the fitted function to the data?¶
Yes. Add the parameter I0 from the Instrument sub-menu to scale the fit; fitting this parameter will autoscale the fit to the data.
I want to use different instruments instances to fit data sets collected of different instruments¶
Multiple instrument can defined in the instrument definition dialog. These could be, for example, x-ray and neutron instruments or the same instrument with different polarization. By double clicking on the simulation function definition in the simulation tab the instrument to use for that particular simulation can be chosen.
In some cases (e.g. polarized neutrons) it may be easier to switch the polarization between datasets instead of using multiple instrument configurations as this ensures all instruments have the same parameters for all other settings.
Note
When ever you change parameters between datasets you have to make sure they are reset for the first dataset. You may not recognize it upon simulation but the fitting algorithm does not re-run the project definition and thus the first dataset will use the same settings as the last.